OPEN SCIENCE
We are strong advocates of open access publications and open source as a development model to promote universal access to our tools and share them with the scientific community. Our work results as developed tools such as fish transgenic lines, DNA constructs or code that we openly share with all the community upon request.
In this line, we actively
- share the code in public repositories as GitHub https://gist.github.com/cristinapujades, where code can be freely used and modified;
- submit our unpublished results to the bioRxiv (https://www.biorxiv.org/), the preprint server for biology;
- publish our raw data in the Dataverse/CORA repository (https://dataverse.org/), the open source research data repository;
- provide information to ZFIN platform, which gathers all published information related to zebrafish to keep the zebrafish community updated with the available resources (https://zfin.org).
DAMAKER: The 3D Digital Atlas MAKER
The 3D-anatomical reconstruction of the tissues’ growth and cell lineage over time requires huge computing power for the visualisation and analysis of the data volumes. If our goal is to extract meaningful biological information from large imaging datasets it is required to develop user-friendly tools to be paired with imaging techniques.
We have developed computationally efficient and robust methods to build a digital representation of the developing vertebrate brain from 3D+time imaging data for further quantitative analysis and multilevel modeling. The 3D digital atlas maker (DAMAKER) is an image analysis pipeline that allows generation of quantifiable 3D-models from confocal images. The 3D-models can be further used to establish a tissue wide 3D atlas of virtually any tissue. It is leveraging registration and segmentation plugins in FIJI as well as homemade macros in order to make the generation of models as reproducible as possible and their quantification as user friendly as possible. Macros allow for the automatic processing of batches of samples at each step. The different steps of the pipeline have been kept independent in order to allow each user to adapt the pipeline to their tissue and application (Blanc et al, eLife 2022).
For further information contact matthias.blanc@upf.edu or cristina.pujades@upf.edu
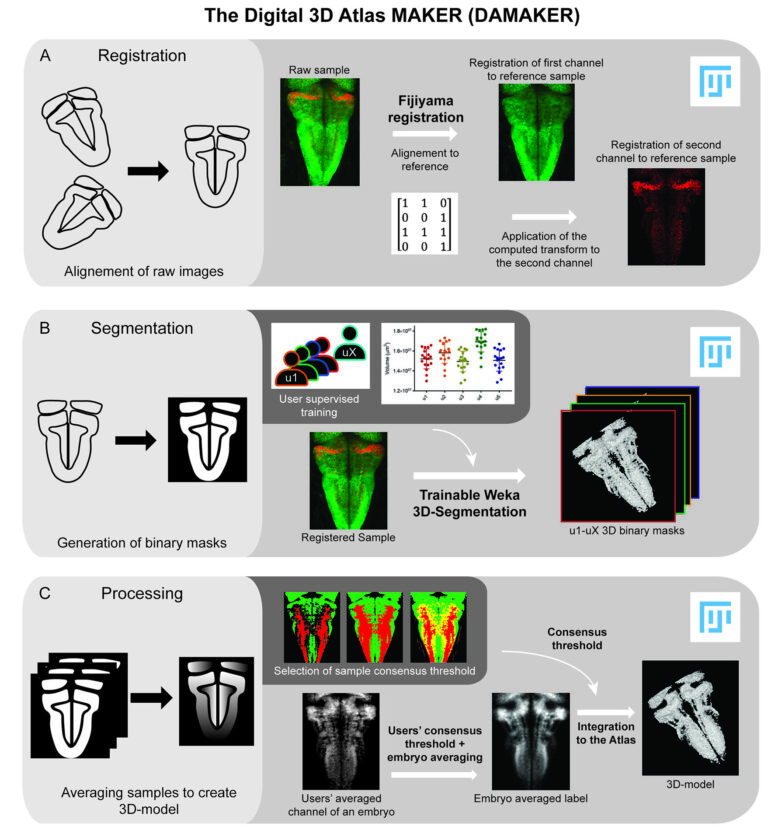
IMAGE PROCESSING TOOLS
Live imaging data – and particularly SPIM data – can be very large and difficult to process. We have found that rather than working with a single large file, it is convenient to work with sets of files. Let’s say for a movie with 400 time steps and two channels, you would create a single file with a unique name for each channel and time point. You can then iterate over these files, concatenate all of them or a subset (x, y, z), process them in batch, save them etc. This enables you to i) work with your data more efficiently by developing your processing pipeline on a reduced representative set of your data, for example every 10th time point or just the region of interest, and ii) save space by saving a processing pipeline, that you can reproduce any time, rather than a lot of heavy processed data.
We have created a small ImageJ/FIJI script that allows you to:
- create a set of files with specific naming from your large microscopy file;
- register your movie in a semiautomated manner (see movie N : CB_18H) you choose a fixpoint that you would like to be always in the same location (x, y, z) in your destination files. You are guided through the frames of the movie and click on that point for each frame. The script then generates the image files with these transformations. Apart from compensating for morphogenetic movements, this operation will likely allow for cropping the data in x, y, z and can save you significant amounts of disk space;
- compensate for fluorescence bleaching or lighting up of transgenic lines;
- generate movies of the process you want to show by extracting only the z plane of interest or a small volume for a maximum intensity projection (see movie M);
- iterate over your files and do any wished processing.